The following FAQ is intended to guide you through the new registration procedure with the FAMHP as an operator in the medical devices sector.
General questions
The different possible activities are defined below. You will find them in the online applications when you register.
These activity definitions are based on the European definitions listed in the European regulations : 2017/745 for medical devices and 2017/746 for in vitro diagnostic medical devices.
You may combine different activities. For example, you may act both as a manufacturer for the devices that you place on the market in your own name and as a distributor for devices placed on the market in the name of a third party.
In order to know whether or not you should register in our online applications, please be sure to state clearly which activities you undertake, according to the definitions below.
1. Manufacturer of medical devices
The definition of manufacturer is mentioned in Article 2, 30 of the Regulation (EU) 2017/745 on medical devices and Article 2, 23 of the Regulation (EU) 2017/746 on in vitro diagnostic medical devices :
« manufacturer »: means a natural or legal person who manufactures or fully refurbishes a device or has a device designed, manufactured or fully refurbished, and markets that device under its name or trademark.
A manufacturer is an operator who places devices on the market under his own name, whether manufactured by himself or by a third party.
A manufacturer is therefore not considered to be a distributor if he makes his own products available on the market.
If you manufacture custom-made devices please refer to the manufacturer of custom-made devices activity.
2. Manufacturer of custom-made devices
The definition of manufacturer is mentioned in Article 2, 30 of the Regulation (EU) 2017/745 on medical devices and Article 2, 23 of the Regulation (EU) 2017/746 on in vitro diagnostic medical devices :
« manufacturer »: means a natural or legal person who manufactures or fully refurbishes a device or has a device designed, manufactured or fully refurbished, and markets that device under its name or trademark.
A manufacturer is an operator who places devices on the market under his own name, whether manufactured by himself or by a third party.
A manufacturer is therefore not considered to be a distributor if he makes his own products available on the market.
The definition of custom-made is mentioned in Article 2, 3 of the Regulation (EU) 2017/745 on medical devices :
« custom-made »: means any device specifically made in accordance with a written prescription of any person authorised by national law by virtue of that person's professional qualifications which gives, under that person's responsibility, specific design characteristics, and is intended for the sole use of a particular patient exclusively to meet their individual conditions and needs.
3. Systems and procedure packs producer
Is considered as a systems and procedure packs producer (Article 22 of Regulation (EU) 2017/745) :
Any natural or legal person who combines devices bearing a CE marking with the following other devices or products, in a manner that is compatible with the intended purpose of the devices or other products and within the limits of use specified by their manufacturers, in order to place them on the market as a system or procedure pack:
(a) other devices bearing the CE marking ;
(b) in vitro diagnostic medical devices bearing the CE marking in conformity with Regulation (EU) 2017/746.
(c) other products which are in conformity with legislation that applies to those products only where they are used within a medical procedure or their presence in the system or procedure pack is otherwise justified.
4. Authorised representative of a manufacturer of medical devices
The definition of authorised representative is mentioned in Article 2, 32 of the Regulation (EU) 2017/745 on medical devices and Article 2, 25 of the Regulation (EU) 2017/746 on in vitro diagnostic medical devices :
« authorised representative »: means any natural or legal person established within the Union who has received and accepted a written mandate from a manufacturer, located outside the Union, to act on the manufacturer's behalf in relation to specified tasks with regard to the latter's obligations under this Regulation.
An authorised representative acts in the name of the manufacturer under the terms of the contract that binds them and must therefore follow the same instructions as are given to the manufacturer.
An authorised representative is considered to be a distributor if he distributes the devices that he represents.
An authorised representative is considered to be a importer if he imports the devices that he represents.
If you import and make available medical devices in respect of which you operate as an authorised representative, please refer to the activity of importer or distributor of medical devices.
5. Distributor of medical devices
The definition of distributor is mentioned in Article 2, 34 of the Regulation (EU) 2017/745 on medical devices and Article 2, 27 of the Regulation (EU) 2017/746 on in vitro diagnostic medical devices :
3° « distributor »: means any natural or legal person in the supply chain, other than the manufacturer or the importer, that makes a device available on the market, up until the point of putting into service.
A distributor is therefore an operator, other than the manufacturer and the importer, who makes medical devices available on the European Union market.
If you place medical devices on the market manufactured by you, or medical devices placed on the market in your own name, please refer to the activity of manufacturer of medical devices.
Note: the retailer is considered as a distributor.:
6. Exporter of medical devices
For medical devices the notion of "exporter" has been abrogated by the Royal Decree of 28 April 2021.
Registration as an exporter is no longer mandatory. However, it is still possible to register on the web portal as an exporter, which gives access to the application « my free sales certificates » which allows the request of electronic free sales certificates for devices placed on the market under the directives.
For devices placed on the market under Regulations (EU) 2017/745 and 2017/746 only manufacturers and authorised representatives can apply for certificates of free sale.
7. Importer of medical devices
The definition of importer is mentioned in Article 2, 33 of the Regulation (EU) 2017/745 on medical devices and Article 2, 26 of the Regulation (EU) 2017/746 on in vitro diagnostic medical devices :
« importer »: means any natural or legal person established within the Union that places a device from a third country on the Union market.
An importer is therefore an operator who makes available medical devices from a country outside the European Union.
8. Health institution
The definition of health institution is mentioned in Article 2, 36 of the Regulation (EU) 2017/745 on medical devices and Article 2, 29 of the Regulation (EU) 2017/746 on in vitro diagnostic medical devices :
« Health institution » means an organisation the primary purpose of which is the care or treatment of patients or the promotion of public health.
Healthcare institutions have access to the web portal to register their in-house device manufacturing or single-use device reprocessing activities and the associated medical devices
9. Service and technical home assistance (STHA)
Are considered STHA (Article 59 of the law on medical devices)
Businesses that, in the course of medical treatment of a patient outside a hospital, install and/or maintain medical devices.
The registration can be done through our web portal
User manuals are available on this portal in order to guide you in registering your company:
Companies based outside Belgium
In the My activity application, you must register under your own activity/-ies. For your registration with the FAMHP to be complete you must notify at least one activity. Your company will then be automatically assigned an FAMHP number.
In the application, the « + » next to each activity allows to add this activity and to open the relevant form.
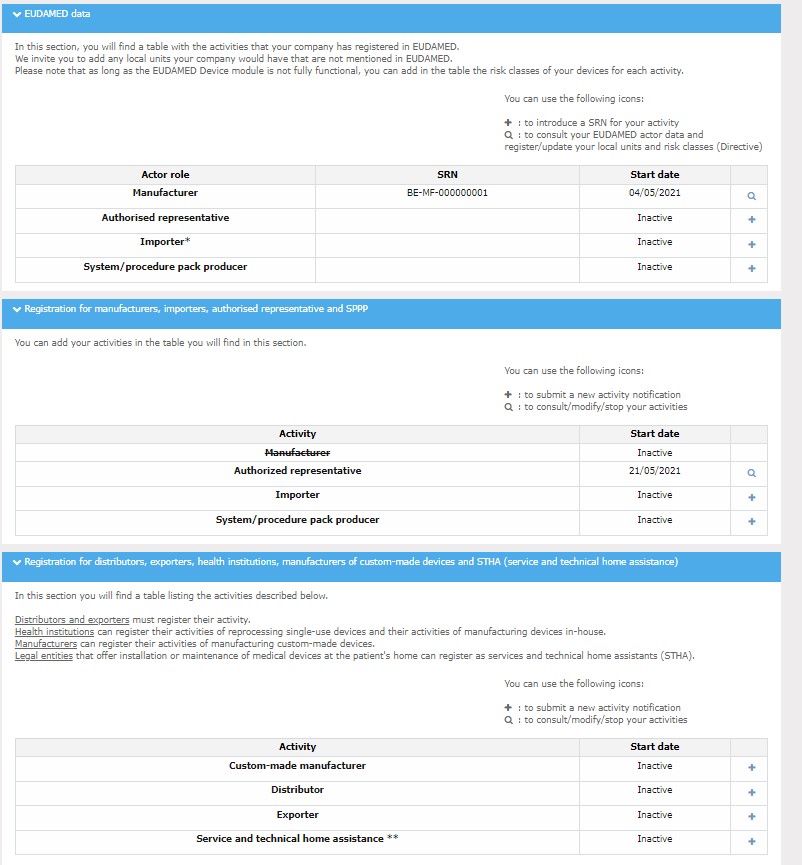
Once the activity notification form is open, you must complete the various information requested:

Once the form is completed, click on "Submit Notification".
We remain at your disposal for
any technical questions, please contact support.portal@afmps.be
any other questions, please contact notifications.meddev@fagg-afmps.be
As a distributor, you will also be asked to fill in the information concerning the manufacturers and authorised representatives for the medical devices that you make available. This information must be registered in the « My adress book » application.
For each activity, you are invited to fill in an autocontrol form (self-monitoring form) in the « My control » application. This form can be used to perform a risk assessment. The « My control » application also enables you to communicate better with the inspector.
This self-inspection form is compulsory for distributors and exporters.
In accordance with Article 6 of the Royal Decree of April 7, 2019 implementing article 35septies of the law on compulsory health care and compensation insurance, coordinated on July 14, 1994, coordinated on 14 July 1994, related to the notification of implants and invasive medical devices for long-term use, the applicant must provide the notification number(s) allocated by the Federal Agency for Medicines and Health Products to distributors of medical devices.
The applicant is defined in Article 1, 5° of this same decree:
5° « applicant »: the company placing or having placed an implant on the Belgian market.
This means that the applicant must make himself known to the FAMHP in order to claim reimbursement for the medical devices that he makes available. Manufacturers, importers and distributor of invasive long-term medical devices and implants who are requesting reimbursement from RIZIV/INAMI must therefore register in the online applications and notify the relevant medical devices in the web portal. You can find out which medical devices fall under this on the website of the RIZIV, notificatieprocedure.
We invite you to register voluntarily, even if you are not legally obliged to do so, in order to receive swift, targeted communications in connection with your registered activities. These communications may concern the future applications that we are developing, information on future regulation, topical trainings …
This registration will improve and speed up administrative procedures with the FAMHP.
Moreover, your registration will facilitate management of your inspections in the My control application. The frequency of these inspections is based on a risk analysis that takes into account various parameters such as registration in our applications and compliance with the guidance documents written by the sector that represents the activity. My control is also a tool for communication between you and the inspector responsible for your inspection.
Of course! You can still sign up on the FAMHP web portal, even if your company is based outside Belgium. By registering, you will be kept informed of FAMHP activities concerning medical devices.
Our applications are currently only available for operators based in the European Union. They are in the process of being revised so that operators based in a third country (outside the European Union), can access them.
Technical questions
This means you didn’t enter your activities (manufacturer, distributor, etc.) in the « My activities » tile. Please log in to the portal and update your activities.
To access Belgian federal applications, a Non-Belgian citizen will need a NISS number, which is a Belgian Social Security Identification Number. This number starts with your year of birth, month of birth + 40, day of birth and ends with 5 figures. You can apply for this number through the Limosa service.
Make sure you have access to the « Authentic source of medical devices » (« Source authentique des dispositifs médicaux ») as described in 3.2.2 (Figure 15).
Registration emails are sent electronically to the legal managers of your enterprise. If they have not received this information, please ask them to check their junk mail folder.
Please check the junk mail folder in your mailbox.
If it’s not there, please request a new password by following the steps in point 4.1 and clicking « Forgot Password » Figure 19).
I have a new phone. How do I get my otp code on it?
Use the link « Forgot Password » on the login page with your email address
You should receive an email containing a link. This link can only be clicked once within 5 minutes. Via this link you can scan QR-code with your new phone.
You can click « Forgot Password » without any restriction.
This person must take steps to obtain a NISS number from the Limosa service.
Once this number has been allocated a, this should be added them to the list of users on CSAM by following the steps in point 3.3.2. An automatic mail with the login and temporary password should be sent.
Make sure the designation of « Institution in the healthcare domain » (« Institution liée au domaine de la santé ») is active (see 3.2.1) and then follow the steps in 3.2.2.
Please see this link.
No, it is not possible. Only a Belgian identity card can be used.
This procedure takes around 15 days.
This number has been sent by an automatic mail.
Uninstall the TOTP login and authenticator app.
Either reinstall another authenticator app (Google Authenticator/Microsoft Authenticator/Authy, etc.); Then synchronise the time zones. Google Authenticator example: access the main menu for the Google Authenticator app -> Settings-> Time correction for codes -> Sync now
Or install the authenticator on another device. Use https://authy.com to install and test it.
Use the Google Chrome or Internet Explorer browser.
There are three possible reasons for this error:
- Could you try logging in again? Sometimes, the social security access authorisation is not sent directly to our apps. You have to wait until the following day to be able to log in.
- Your company is not designated as an « Institution in the domain of healthcare ». (see 3.2.1).
- You are not a named manager or are not flagged as a user of our apps (see 3.2.2)
- If none of the above solutions worked for you, please send us an email with a screen print of the social security page with your access details.
Yes, you will need to register again. The whole actor registration and information procedure has been modified since December 21st 2017. This new procedure is completely free of charge
For any technical question related to the use of the web portal, you can contact the technical support of the portal at the email address support.portal@fagg-afmps.be or by phone at +32 2 528 48 56.